24.jpg)







Introduction
Background
Biliary atresia is a condition in which the normal extrahepatic biliary system is disrupted. Progressive damage of extrahepatic and intrahepatic bile ducts secondary to inflammation may occur, leading to fibrosis, biliary cirrhosis, and eventual liver failure. Although various treatment options are under investigation, the primary therapy is surgical.1,2,3,4
Biliary atresia affects approximately 1 in 10,000-15,000 births and occurs in 2 distinct clinical forms: fetal-embryonic (or syndromic) and perinatal (or acquired).
The fetal-embryonic form is characterized by early cholestasis, appears in the first 2 weeks of life, and accounts for 10-35% of all cases. In this form, the bile ducts are discontinuous at birth, and 10-20% of affected neonates have associated congenital defects, including situs inversus, polysplenia, malrotation, intestinal atresia, and cardiac anomalies, among others. The perinatal form of biliary atresia accounts for the remaining 65-90% cases. This form is typically found in neonates and infants aged 2-8 weeks. Progressive inflammation and obliteration of the extrahepatic bile ducts occurs after birth. This form is not associated with congenital anomalies, and infants may have a short jaundice -free interval.
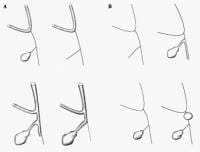
Biliary atresia may be classified according to whether the disease can or cannot be corrected. As Image 1 shows, in the correctable group (10-15% of cases), the proximal common hepatic duct is patent, allowing for primary anastomosis of the extrahepatic bile duct to the bowel. Resection of the fibrous bile duct remnant may be done, followed by a Roux-en-Y anastomosis of the bowel to the bed of the porta hepatis, according to the Kasai portoenterostomy procedure. In the uncorrectable group, the extrahepatic bile ducts do not have the same patency as in the correctable group.
Types of biliary atresia. A, Operable, or correctable, biliary atresia, a major portion of the extrahepatic bile ducts are patent. B, The inoperable group does not have the patency shown in A. Reproduced with permission from BC Decker, 2000.
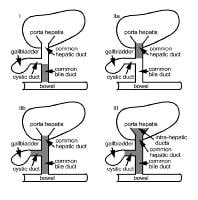
Another method of classification, the Kasai classification system, is widely used and divides cases of biliary atresia according to their location and degree of pathology. As shown in Image 2, 3 main types of biliary atresia are defined:
- In type I, the common bile duct is obliterated while the proximal bile ducts are patent.
- In type II, atresia of the hepatic duct is seen, with cystic bile ducts found at the porta hepatis. In type IIa, the cystic and common bile ducts are patent, whereas in type IIb, the cystic, common bile duct, and hepatic ducts are all obliterated.
- Type III atresia refers to discontinuity of both the right and the left hepatic ducts to the level of the porta hepatis. Unfortunately, type III biliary atresia is common, accounting for more than 90% of cases.
Classification of biliary atresia according to the location of involvement (gray areas). Type I is obliteration of the common bile duct while the proximal bile ducts are patent. Type IIa is atresia of the hepatic duct with cystic bile ducts found at the porta hepatis. Type IIb is atresia of the cystic duct, common bile duct, and hepatic ducts. Type III is involvement of the extrahepatic biliary tree and intrahepatic ducts of the porta hepatis.
Recent studies
In a study by Petersen et al, endoscopic retrograde cholangiopancreatography (ERCP) was performed in cholestatic patients younger than 6 months suspected of having an extrahepatic cause of cholestasis, particularly biliary atresia. In this series, the sensitivity of ERCP for diagnosing biliary atresia was 92% and specificity was 73%. The authors noted that in preselected patients, ERCP is not an alternative to noninvasive imaging but can avoid unnecessary surgical procedures in approximately 25% of cases. They therefore recommended that ERCP be performed before explorative laparotomy in all patients suspected of having biliary atresia.5
In a retrospective analysis by Shanmugam et al, ERCP had both a high positive and a high negative predictive value for biliary atresia in cholestatic infants younger than 100 days. Of 48 patients, ERCP demonstrated a patent biliary tree in 20 patients, and in 3 infants in whom cannulation failed, biliary atresia was confirmed at exploratory laparotomy. The remaining 25 infants, who were diagnosed with biliary atresia by ERCP, also proceeded to exploratory laparotomy, in which biliary atresia was confirmed in 22.6
Pathophysiology
The pathogenesis of biliary atresia is poorly understood. Although several mechanisms are implicated, no single etiologic factor is identified. Association with congenital anomalies in some infants suggests genetic factors. Infection with cytomegalovirus, group C rotavirus, and reovirus type 3 has been implicated in certain cases. Immune-mediated ductal injury is another consideration. Cholestasis almost certainly contributes to ongoing hepatocellular and biliary damage in infants with the disorder.7
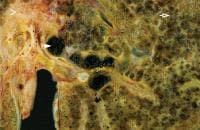
Histologic findings on liver biopsy typically include acute on chronic inflammatory change with obstruction, fibrosis, and the proliferation of ductal and glandular elements. Findings may not be definitive early in the course of the disease, and repeat biopsy may be required to reach a definitive diagnosis.Image 3 depicts the histologic findings associated with biliary atresia.
Histologic findings associated with biliary atresia. Image shows end-stage biliary cirrhosis with micronodules (open arrow). Extensive perivascular fibrosis (arrowhead) and dilated inspissated cystic areas (closed arrow) are seen at the hepatic hilum about 5-10 mm from remnants of the obstructed bile duct. Image courtesy of James Phillips, MD, Department of Pathology, Hospital for Sick Children.
Frequency
United States
The overall incidence is 1 in 10,000-15,000 live births.
International
The incidence is higher in the Asian population than in other groups.
Mortality/Morbidity
Although the exact numbers may vary, if done in the first 2-3 months of life, a hepatoportoenterostomy can restore bile flow from the liver to the bowel in 60-80% of cases. The highest success rates are seen when surgery is performed earlier, before 2 months of age. It is thought that 20-30% of patients who undergo the Kasai procedure demonstrate long-term stability of their disease, although ongoing aggressive nutritional support may be required. Alternatively, liver transplantation may be lifesaving. The overall survival rate for patients after the Kasai procedure, including those who require subsequent liver transplantation, is thought to be more than 80%.
Race
In the United States, African-American infants are more commonly affected than white infants.
Sex
A slight female predominance is noted.
Presentation
Clinical manifestations of biliary atresia are nonspecific. Infants are often born at full term, have a normal gestational history with appropriate initial weight gain, and appear healthy despite having jaundice. Stools tend to become progressively acholic during the first weeks of life. Although hepatosplenomegaly may appear early, chronic hepatic failure is rare at the time of diagnosis. Associated congenital anomalies may or may not be present. Some cases present early with bleeding due to malabsorption and resulting vitamin K deficiency.8,9,10
No single biochemical marker can be used to distinguish biliary atresia from other causes of neonatal cholestasis. Laboratory values often show a moderate conjugated hyperbilirubinemia, a mild to moderate elevation of serum transaminases, and a striking elevation of gamma-glutamyl transferase. The patient's prothrombin time and alkaline phosphatase may be elevated. Conjugated (direct) hyperbilirubinemia is defined by a serum conjugated bilirubin concentration greater than 2 mg/dL (34.2 mmol/mL) or 15% of the total bilirubin level. At the time of diagnosis, the total serum bilirubin level is typically 6-12 mg/dL with 50-80% conjugated.
Neonatal jaundice may be due to a variety of causes, including hemolysis, sepsis, and metabolic disease. When jaundice persists longer than 14 days, biliary atresia and idiopathic neonatal hepatitis must be considered. Together, these conditions cause more than 90% of all cases of neonatal obstructive cholestasis.
Early differentiation of extrahepatic biliary atresia from medical causes of hepatic dysfunction is important because earlier surgical intervention is directly associated with better outcomes. The North American Society for Pediatric Gastroenterology, Hepatology and Nutrition guidelines suggest that 2-week old infants with jaundice should be evaluated for cholestasis. If the child is breast-fed, this may be delayed until 3 weeks if the physical examination is normal, there is no evidence of dark urine or light stools, and the child will be carefully monitored.
Evaluation should include a complete history and physical examination, as well as laboratory assessment. In some countries, stool color cards are used to screen infants for biliary atresia; however, the efficacy of this screening tool is still being reviewed, and the specificity is thought to be low. Radiologic studies, including abdominal ultrasound, hepatobiliary scintigraphy, magnetic resonance cholangiopancreatography (MRCP), and endoscopic retrograde cholangiopancreatography (ERCP), may also be helpful.
Preferred Examination
Several imaging modalities have been used in the diagnosis of biliary atresia. Although some findings are highly suggestive of the disease, none is pathognomonic, and reliance on more than one test is common.
Ultrasonography is often the initial investigation in patients with suspected biliary atresia, followed by hepatobiliary scintigraphy, a study that has been used effectively for many years. If the diagnosis remains elusive after ultrasonography and hepatobiliary scintigraphy, magnetic resonance cholangiopancreatography (MRCP) may be helpful. Liver biopsy is often used to confirm the diagnosis of biliary atresia and may be done at the same time as surgical or percutaneous cholangiography. Alternative procedures include duodenal intubation and endoscopic retrograde cholangiopancreatography, which are generally not considered in infants.
Differential Diagnoses
| Alagille Syndrome | Hepatitis A |
| Biliary Disease | Hepatitis B |
| Biliary Obstruction | Hepatitis C |
| Cholelithiasis | Rubella |
| Cystic Fibrosis | |
| Cytomegalovirus Infection |
Other Problems to Be Considered
Extrahepatic causes of neonatal cholestasis: eg, biliary atresia, bile duct stenosis/ stricture/ or cholelithiasis, tumors or masses, choledochal cyst.
Intrahepatic causes of neonatal cholestasis: infection (eg, CMV, rubella), metabolic (cystic fibrosis), genetic (eg, Alagille syndrome), toxicity (eg, aluminum, TPN associated), other (eg, idiopathic neonatal hepatitis).
Alagille syndrome
Turner syndrome
Trisomy 21
Caroli's disease
Progressive familial intrahepatic cholestasis
Cystic fibrosis
Alpha-1-anti-trypsin deficiency
Galactose-1-phosphate uridyltransferase deficiency (galactosemia)
Gaucher's disease
Wolman's disease
Neonatal Dubin-Johnson syndrome
Hypothyroidism
Panhypopituitarism
Inborn errors of bile acid synthesis
Cytomegalovirus infection
Herpes simplex virus infection
Rubella
Syphilis
Toxoplasmosis
Bacterial sepsis
Erythroblastosis fetalis
Choledochal cyst
Idiopathic neonatal hepatitis
Nonsyndromic intrahepatic bile duct hypoplasia
Total parenteral nutrition (TPN)–associated cholestasis...
More on Biliary Atresia |
 Overview: Biliary Atresia Overview: Biliary Atresia |
| Imaging: Biliary Atresia |
| Follow-up: Biliary Atresia |
| Multimedia: Biliary Atresia |
| References |
| Further Reading |
| Next Page » |















No comments:
Post a Comment